9 Immune-mediate myopathies
Generally speaking, immune-mediated disease is the result of abnormal immune response against self-peptides or antigens.
A difficulty when evaluating immune-mediate disease is determining whether the immune response is the primary cause of muscle damage, or whether the leukocytes are simply responding to the damage. In the case of immune-mediate myopathies, the invasion of intact myofibers by mononuclear leukocytes is characteristic.
9.1 Masticatory myositis
This is a rare condition of dogs characterized by profound atrophy of the muscles of mastication: the masseter, temporal, and pterygoid muscles. Two conditions, atrophic myositis and eosinophilic myositis, formerly thought to be distinct entities, are now known to represent two manifestations of the single disease known as masticatory myositis.
Animals present with difficulty opening their mouth and concurrent muscle atrophy. Pain is noted upon opening the mouth, and the jaw remains difficult to open even under general anesthesia. German shepherds seem to be predisposed, but the condition affects a variety of breeds.
The root cause of masticatory myositis is a unique myosin isoform, 2M myosin, found only in the masticatory muscles of the dog. Antibodies against 2M myosin lead to a patchy lymphocytic inflammatory response, ultimately resulting in variably severe, multifocal, polyphasic necrosis. Both T- and B-cells are present in the inflammatory response. In some cases, eosinophils are present in large numbers. Prompt treatment with corticosteroids can resolve clinical signs and lead to recovery. Untreated cases result in necrosis followed by fibrosis, which, if severe, can be devastating. A serologic test to detect antibodies against 2M myosin is available.
9.2 Polymyositis of dogs
Immune-mediate polymyositis is generalized myopathy of dogs. It can mimic masticatory myositis, and both should be considered as differential diagnoses when confronted with a dog with masticatory muscle atrophy. The condition occurs in adult dogs of a variety of breeds, though German shepherds are again overrepresented. Clinical signs are variable and include muscle atrophy, exercise intolerance, weakness, stiff gait, and pain on deep muscular palpation. Muscle atrophy may be mild to severe and is generalized. The esophagus may be affected. Dogs with immune-mediate polymyositis generally do not have antibodies against 2M myosin. The disease is mediated by CD8+ T-cells, with little to no involvement of B cells, a feature that helps differentiate this condition from masticatory myositis. Muscle necrosis multifocal and polyphasic, myofibers are frequently invaded by lymphocytes, and there is evidence of degeneration, regeneration, and fibrosis.
9.3 Acquired myasthenia gravis
Myasthenia gravis is a rare but important disease of dogs and cats (and humans), and two clearly defined types exist: acquired and congenital. Acquired myasthenia gravis is caused by antibodies against the acetylcholine receptors located at the motor end plate (Figure 9.1). Binding of antibodies to the receptors forms an immune complex that results in endocytosis, ultimately decreasing the density of acetylcholine receptors at the neuromuscular junction. Lack of Ach-R in turn reduces the ability of the myofiber to depolarize, resulting in the clinical signs noted below. In dogs, the condition has been linked to thymomas, which are thought to create an abnormal immune response leading to antibody formation. It has also been linked to hypothyroidism and various malignant neoplasms.
A variety of breeds are affected, and age of onset follows a bimodal distribution, with one peak at 3 years and another at 10. Affected dogs can present with one of three clinical syndromes:
- Generalized: these animals have generalized weakness, suffer from exercise induced collapse, and commonly regurgitate due to megaesophagus. Aspiration pneumonia may result from regurgitation.
- Localized: this form preferentially involves the esophageal, facial, and pharyngeal muscles, leading to megaesophagus and regurgitation, but no weakness.
- Fulminant: rapid and sustained generalized weakness.
Gross lesions of acquired myasthenia gravis are limited to disuse atrophy, megaesophagus, and possibly aspiration pneumonia or thymoma. Microscopic lesions are underwhelming, but immune complexes at the neuromuscular junction can be demonstrated using immunohistochemistry. Detection of circulating anti-acetylcholine-receptor antibodies is the (antemortem) diagnostic test of choice.
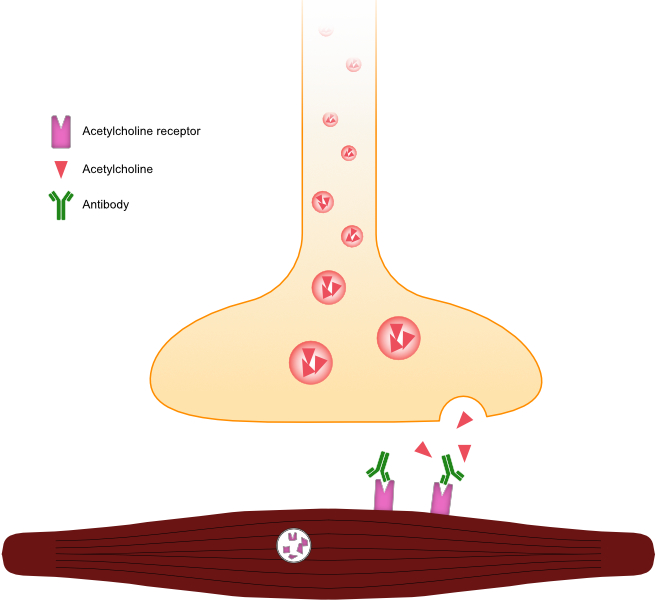
Figure 9.1: Illustration of a motor end plate affected by acquired myasthenia gravis. Note the immunoglobulin binding to the acetylcholine receptor, preventing the binding of acetylcholine, and also leading to endocytosis and decreased receptor density.